- 网页首站
- 企业展示
- 产品中心
- 新闻动态
- 合作伙伴
- 招贤纳士
- 联系我们
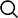

SLAM-seq—RNA 合成及降解代谢动力学分析,基因动态表达研究的理想选择
基因表达是一个动态的过程,取决于细胞内环境的稳态以及对环境的适应性,基因信息调控的转变可能会导致人类疾病的发生。这些基本生物学进程的背后是一个非常精密的分子调控事件,它们控制着 RNA 转录,加工和降解相关的动力学(图 1)。了解基因调控的分子基础需要以转录特异性和系统的方式深入了解 RNA 合成和降解相关动力学。但是,对于常规的 RNAseq,我们只能获得终点水平的 RNAs 丰度信息,无法区分及定量新合成的新生 RNAs 水平,因此,常规的 RNAseq 不能对 RNA 的合成和降解相关的动力学进行分析。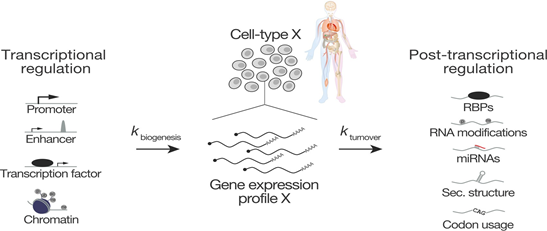
图 1 mRNA 水平由转录、加工处理以及降解所决定
RNA 动力学研究
为了对 RNA 的动力学进行分析,研究者们就开发了多种方法来分析新合成的(nascent)RNA;这些方法揭示了在启动子处的差异转录程度,表明 RNA 聚合酶 II(Pol II) 在启动子附近的暂停是基因表达的关键调节步骤,证明了 nascent RNA 有直接调节转录的作用,并表明其序列和结构影响转录的延伸、暂停和停顿,以及发挥染色体修饰结合和增强子的作用。nascent RNA- seq 方法旨在区分新近转录的 RNA 和其它 RNAs,这些方法可以分为 4 类:溴尿苷 (BrU) 免疫共沉淀深度测序分析(BRIC-seq),4sU-seq,瞬时转录组测序(TT-seq),代谢标记法(SLAM-seq)(图 2)。
 图 2. RNA 动力学研究方法
图 2. RNA 动力学研究方法
RNA 动力学研究不同方法的优缺点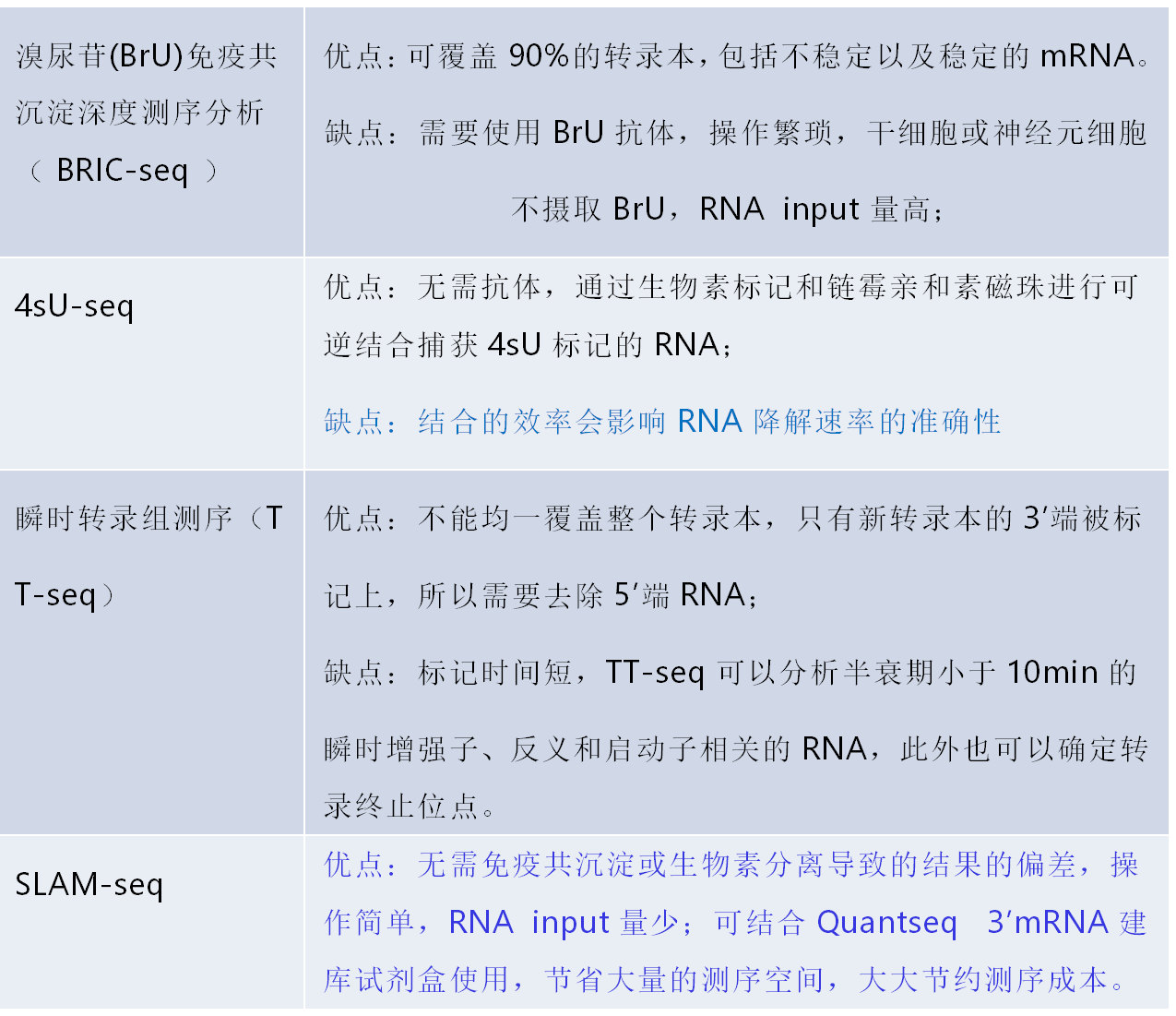
图 3 SLAMseq 工作流示意图相对于其他的方法,SLAM-seq(图 3)表现超强的优势,无需繁琐且技术要求苛刻的免疫共沉淀或生物素分离等操作,操作简单,RNA input 量少;可结合 Quantseq 3’mRNA 建库试剂盒使用,节省大量的测序空间,大大节约测序成本,使其成为 RNA 代谢动力学研究的优质选择,同时,SLAM-seq 技术方法学发表在了 Nature Methods 上。
培养的细胞用 4-硫代尿苷(S4U) 标记新生 RNA(绿色)。抽提纯化总 RNA,并加入 IAA 诱导 4-巯基烷基化。用 QuantSeq 3』 mRNA-Seq 文库构建试剂盒进行文库构建,在反转录过程中,S4U 位点会反转录成鸟嘌呤 (G,红色) 而不是腺嘌呤 (A,黑色)。因此,在随后的数据分析过程中,可以通过 T>C 突变信息区分已有的 RNA 和新合成的 RNA。
SLAm-seq 可对新合成(s4U-标记)的转录本进行定量
用 100μM (毒性浓度远低于 EC50 值)的 s4U 对小鼠的胚胎干细胞(mESC)进行标记 (图 4)。RNA 代谢标记 24 h 后,提取总 RNA,并进行烷基化处理以及 3 '端 mRNA 测序 (Quant-seq)。Quantseq 可对含 poly A 结构的 RNA 生成靠近 3』端序列的文库,每个转录本只产生一个文库片段,可对 mRNA 进行准确的定量分析。此外,3 '端测序可对特定细胞类型的非编码区域(UTR)进行重新评估注释,从而对 mRNA 3』 Isoform 特意性表达分析。在 s4U 代谢标记 24 小时后,mESCs 通过 Quant-seq 流程生成 SLAM seq 文库,与未标记条件的比较,可观察很高到 T >C 转换累积 (图 4b)。全转录组分析证实了这一观察 (图 4c)。在没有 s4U 代谢标记的情况下,我们观察到对于任何给定的转换,中位数比率≤0.1%,这与 Illumina 报道的测序错误一致,而 s4U 标记后具有统计学意义 (P <10−4,Mann-Whitney 检验),T < C 转换率增加 50 倍 (图 4c),在被覆盖基因组区域均匀分布 (图 4)。值得注意的是,非 T > C 转化率仍然低于预期的测序错误率 (图 4c),在没有代谢标记的情况下,用 IAA 处理总 RNA 并不影响定量基因表达分析。

图 4 SLAm-seq 对 s4U-标记的转录本进行定量
SLAm-seq 可评估增强子的活性
在丰度相对稳定的,具有可比性的转录本中(~100 cpm),T>C reads 数在特定转录本之间存在差异(图 5a)。ESC 特异性转录因子 Sox2 以及 pri-miR-290-295,T>C reads 数水平高,而管家基因 GDPH T>C reads 水平很低。这可能是因为其高稳态表达水平是通过转录本高稳定性的积累来实现的。
在 ESCs 中,多能状态主要由少量增强子相关的主要转录因子控制,包括 Oct4、Sox2 和 Nanog,它们驱动维持 ESC 状态所必需的靶基因的表达。结果显示,在 s4U-标记实验中,Oct4、Sox2 和 Nanog (OSN) 调控的靶基因在 T > C reads 中有很高的水平(图 5b)。从全局来看,与 4,994 个近端不含 OSN 增强子的基因相比,含 OSN 增强子的 2029 个基因(>5 cpm)的转录本产生的 T> Creads 显著增高 (Mann–Whitney test, P < 10−4; 图 5c)。此外,在靠近强增强子(strongenhancer, SE)的 156 个基因显示出更高的转录水平 (Mann–Whitneytest, P < 10−4; 图 5c)。因此,SLAMseq 可以通过定量转录本的表达来评估增强子的活性。

图 5 mESCs 中多聚腺苷酸转录本的丰度水平
SLAm-seq 提高基因差异表达检测的灵敏度
SLAMseq 可以通过区分已有的 RNAs 以及新合成的 RNAs 来扣除背景信号,可通过对药物处理后新合成的 RNAs 进行差异分析,提高基因差异表达检测的灵敏度(图 6)。
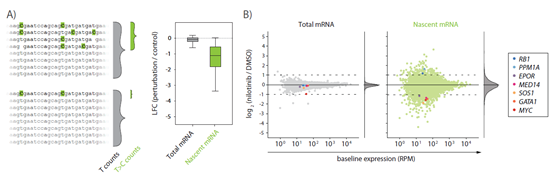
图 6 SLAMseq 提高基因差异表达检测的灵敏度。A) BCR / ABL 抑制剂处理组(尼洛替尼)与 DMSO 对照组相比,K562 细胞新合成 RNA(T>C转换reads,绿色显示)水平显示出比总 mRNA(灰色)更高的变化倍数。B)新合成 mRNA(绿色)与总 mRNA 水平(灰色)对比分析,前者检测到更多的差异表达基因。图片来源于 Muhar et al, 2018.
SLAm-seq 可对转录本稳定性进行检测
以往的研究表明,特定 mRNA 的半衰期与其生理功能密切相关。对检测准确度高的(r2 > 0.6)6665 个转录本的半衰期进行了排序,并对 666 个稳定或不稳定的 mRNA 进行了富集分析。发现 RNA 聚合酶 II 依赖性转录调节因子,这些转录本的半衰期短,然后是翻译,呼吸电子传递,最后是氧化磷酸化(图 7)。
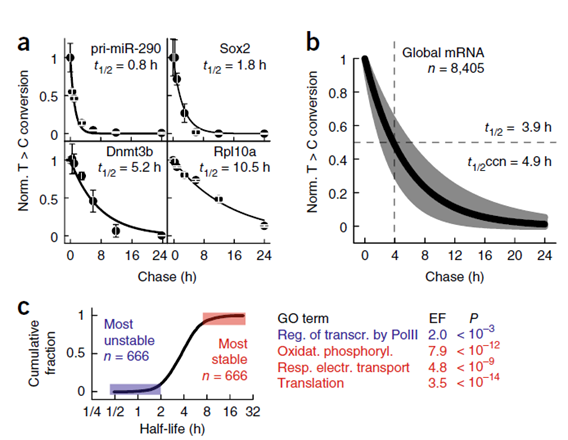
图 7 mESCs 中整体以及特异性转录 mRNA 的稳定性。
SLAm-seq 可揭示 mRNA 稳定性的分子决定因素
在分子水平上,mircoRNA 是核糖核酸蛋白复合物的向导,与靶向 mRNA 的 3』UTR 互补位点相结合。microRNAs 通过抑制翻译和/或促进 mRNA 的降解来诱发其功能。通过对转录本是否含有 microRNA 以及 m6A 靶点进行半衰期分析,发现含 microRNA 靶点以及 m6A 靶点的转录本半衰期显著小于无靶点的转录本(图 8)。
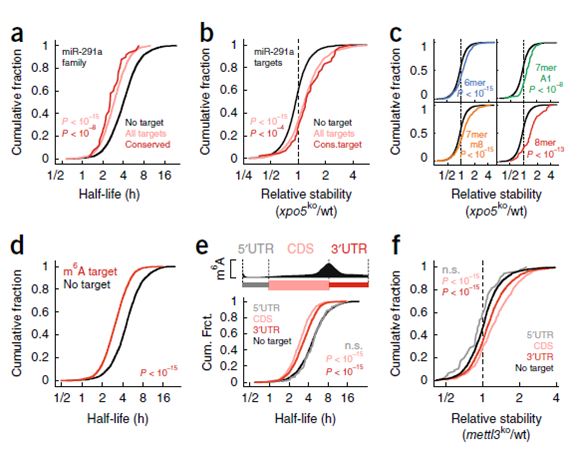
图 8 mESCs 中 mRNA 稳定性的分子决定因素。
在生物学和基础医学研究领域,技术革新总能够带来新的研究思路,推动学科的发展。SLAM-seq 做为一种新的测序方式,在为 RNA 的研究提供了新的视角和思路。未来可以预见,SLAMseq 这一强大的手段能够为生物学以及基础医学研究带来更多的福音。
参考文献:
Herzog, Veronika A.; Reichholf, Brian; Neumann, Tobias; Rescheneder, Philipp; Bhat, Pooja; Burkard, ThomasR.; …; Zuber, Johannes; Ameres, Stefan L. (2017): Thiollinked alkylation of RNA to assess expression dynamics. NatureMethods. DOI:10.1038/nmeth.4435.
Muhar, Matthias;Ebert,Anja; Neumann, Tobias; Umkehrer, Christian; Jude, Julian; Wieshofer, Corinna; …; Ameres,StefanL.; Zuber,Johannes.(2018):SLAM-seqdefinesdirectgene-regulatoryfunctionsofthe BRD4-MYCaxis.Science. DOI:10.1126/science.aao2793.
